For over 40 years, RAS genes have been a major headache for cancer researchers. They were first identified as drivers of cancer back in 1982, and since then, they’ve been linked to a ton of different cancers.
But no matter how much we’ve learned about them, RAS has always been tough to crack when it comes to treatments. It earned the nickname “undruggable” because of all the failed trials and the tricky biology behind it.
Now, though, there’s a new therapy that seems to work by targeting a key step after RAS in the cancer process, and it’s a big deal.
To really grasp why this is so important, it helps to first dive into how RAS works, how it messes with cells, and why so many treatments have fallen short until now.
Introduction to RAS: The Cell’s Master Growth Switch
The RAS gene family has three main members: KRAS, NRAS, and HRAS, and they produce proteins that are key to how cells normally work.
These proteins are small enzymes called GTPases, which act like switches, flipping between an “on” and “off” position to control a bunch of important cell functions. In a healthy cell, RAS proteins help manage things like growth, survival, and division.
Their function is governed by a tightly regulated cycle of binding to one of two guanine nucleotides: guanosine triphosphate (GTP) or guanosine diphosphate (GDP).
When bound to GTP, the RAS protein is in its active, “on” state, capable of binding to and activating downstream effector proteins that propagate growth signals. This activation is typically initiated by external signals, such as growth factors binding to receptors on the cell surface.
The process is facilitated by proteins known as guanine nucleotide exchange factors (GEFs), which promote the release of GDP and the binding of the more abundant GTP. To turn the signal “off,” GTPase-activating proteins (GAPs) bind to RAS and dramatically accelerate its intrinsic ability to hydrolyze GTP back to GDP, returning the protein to its inactive state.
This “on-off” cycling makes sure that growth signals are sent only when needed and stopped when they’re no longer necessary, keeping everything in balance.
The Corrupted Switch: How RAS Mutations Drive Cancer
RAS plays such a big role in cancer because certain mutations mess up its ability to turn off properly. Most of these are small changes. Just a single “letter” swapped in the DNA, that tweak the protein in a way that breaks its shut-off function. These changes usually show up at three specific spots in the gene: codons 12, 13, or 61.
Normally, RAS can turn itself off by breaking down GTP, but with these mutations, that ability is basically gone. The result? RAS gets stuck in the “on” position, constantly sending signals that tell the cell to grow and divide, even when it shouldn’t. That’s one of the defining traits of cancer: uncontrolled growth.
What’s wild is how common these mutations are. They show up in somewhere between 19% and 30% of all cancers, making RAS the most frequently mutated oncogene we know of.
And when it’s mutated, it’s usually bad news. These cancers tend to be more aggressive, harder to treat, and linked with worse outcomes for patients.
The “Undruggable” Target: A 40-Year Challenge

Even though scientists identified RAS as a promising cancer target back in the early ’80s, trying to actually drug it turned into a decades-long failure. For 40 years, nothing worked. The whole field started calling RAS “undruggable” because the biology just didn’t cooperate.
There were two big problems. First, RAS binds to molecules like GTP and GDP so tightly, at levels that are almost impossible to compete with. Plus, GTP is everywhere inside cells, so trying to knock it off with a drug was like trying to empty the ocean with a spoon.
Second, RAS doesn’t have the usual deep pockets or grooves on its surface that small-molecule drugs need to latch onto. The protein is smooth, featureless, and just not drug-friendly. There was no obvious place for a drug to fit.
So researchers tried working around it. One early approach was using farnesyltransferase inhibitors, or FTIs. The idea was to block a step RAS needs to anchor itself to the cell membrane, which is where it does its job. It kind of worked for HRAS-driven cancers, but totally flopped for KRAS and NRAS.
Those proteins just used a backup system, another enzyme that attached a different lipid, and kept functioning anyway. That failure hit hard and pushed a lot of companies to walk away from RAS entirely.
But the sheer difficulty of the problem is what eventually sparked a new wave of innovation. The need was too big to ignore. In 2013, the National Cancer Institute launched the RAS Initiative to jumpstart progress and get researchers working together with better tools and shared data.
That, combined with new drug discovery techniques like covalent inhibitors and chemoproteomics (used by companies like Vividion), finally started to break the cycle.
It took years of failure to force a fresh way of thinking, but that pressure is exactly what led to the breakthroughs that proved RAS wasn’t undruggable after all.
Similar Article: Your Brain is Hiding the Reality from You, And That’s Exactly What Keeps You Alive
A Family of Outlaws: The Diverse Roles of KRAS, NRAS, and HRAS
Targeting RAS isn’t as simple as just hitting one thing. The three main RAS proteins, KRAS, NRAS, and HRAS, are different from each other, both in how they mutate and the kinds of cancers they’re linked to. So trying to develop a single treatment for all of them probably won’t work across the board.
KRAS is the undisputed dominant isoform, accounting for approximately 85% of all RAS mutations in cancer. It is the primary driver in three of the most lethal malignancies: pancreatic ductal adenocarcinoma (mutated in over 90% of cases), colorectal cancer (~45%), and non-small cell lung cancer (~30%).
NRAS is the second most common, representing about 11% of RAS mutations. It is most frequently mutated in cutaneous melanoma (~20%) and certain hematologic malignancies like myeloid leukemia.
HRAS is the least frequently mutated isoform, found in only about 4% of RAS-driven cancers. Its mutations are associated with rarer cancers, including those of the bladder, thyroid, and head and neck.
On top of that, each RAS isoform has its own mutation hotspots. For example, KRAS mutations usually happen at codon 12, while NRAS mutations are more likely to show up at codon 61.
Isoform
Prevalence in RAS-Mutant Cancers
Common Associated Cancers
Primary Mutation Hotspots
KRAS
~85%
Pancreatic, Colorectal, Lung (Adenocarcinoma)
Codon 12 (e.g., G12D, G12V, G12C)
NRAS
~11%
Melanoma, Hematologic Malignancies (e.g., Leukemia)
Codon 61 (e.g., Q61R, Q61K)
HRAS
~4%
Bladder, Thyroid, Head & Neck
Codons 12 and 61
Now, there’s been some progress with drugs like sotorasib and adagrasib, which target a very specific KRAS mutation, G12C. This is a huge win for non-small cell lung cancer, where G12C is common.
But for other cancers, like pancreatic cancer, where KRAS mutations like G12D or G12V are more common, these drugs aren’t effective. It’s clear that creating a drug for each mutation would be a massive challenge.
That’s why many researchers are focusing on finding treatments that target a common pathway downstream of all RAS proteins, something that could work across all RAS mutations, regardless of the specific change in the gene.
Also Read: Just Eat These Every Day Like Japan’s Oldest Doctors – Science Says You Could Live to 100
Collateral Damage: The Problem with Targeting Cancer’s Supply Lines
When you can’t directly target the problem in cancer, another strategy is to go after its supply lines. Basically, the pathways that help fuel the cancer’s growth.
For RAS-driven cancers, one of the key pathways to target is PI3K, an enzyme that relays growth signals inside the cell. But here’s the catch: while PI3K helps cancer grow, it’s also absolutely vital for normal bodily functions, making it a tricky target.
PI3K is central to how cells respond to signals, especially from RAS. It activates other proteins, like Akt, which promote cell growth and survival, things mutant RAS hijacks to drive cancer.
Because of this, blocking PI3K seemed like a good therapeutic idea. But there’s a huge problem: PI3K is involved in many essential processes in healthy cells, especially when it comes to metabolism.
The most obvious example is its role in insulin signaling. After you eat, your blood sugar goes up, and your pancreas releases insulin. This triggers PI3K in cells like muscle and fat, which then helps move glucose into cells for energy or storage.
PI3K also helps control blood sugar by promoting the storage of glucose and stopping your liver from making more. Without PI3K, your body would have trouble managing blood sugar.
This brings us to the problem with trying to block PI3K in cancer treatment. Early PI3K inhibitors were designed to shut down the enzyme completely. They worked in lab tests, slowing cancer cell growth, but when used in patients, they caused major side effects.
The main issue is that by blocking PI3K entirely, these drugs also messed up insulin signaling in healthy tissues. This led to dangerously high blood sugar levels, a condition called hyperglycemia. This was so severe that doctors couldn’t give patients enough of the drug to treat the cancer effectively without causing harmful metabolic issues.
The failure of these early drugs highlights a bigger issue in cancer therapy. The first wave of cancer treatments used broad, non-specific drugs that killed fast-growing cells, cancerous or not. Then came targeted therapies, which aimed at specific proteins overactive in cancer cells.
But as the PI3K story shows, even targeted drugs can fail if the protein they target is essential for normal function in healthy cells.
This is why we need a new kind of therapy, one that doesn’t just block a protein, but precisely targets the problem in cancer cells, leaving the healthy functions of the protein intact. That’s the next frontier in cancer treatment.
Also Check: According to Cognitive Science, This Is the Most Powerful Way to Learn Faster and Improve Your Memory
A Precision Strike: A New Paradigm for Drug Action

When directly targeting a cancer’s main driver doesn’t work, the next best approach is to cut off its supply lines, or the pathways that keep it growing.
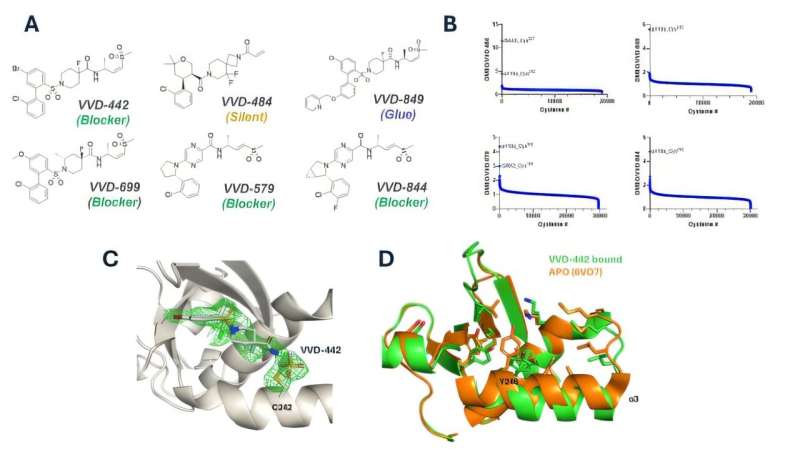
But instead of shutting down a whole pathway, researchers at the Francis Crick Institute and Vividion Therapeutics came up with a much smarter, more precise strategy. Instead of trying to destroy the target, they designed a molecule to specifically disrupt one critical interaction driving the cancer. And the results from preclinical tests are promising.
The key breakthrough is this: rather than trying to block RAS directly or shut down the PI3K pathway entirely, the researchers aimed to stop RAS and PI3K from interacting.
In RAS-driven cancers, the constant “handshake” between RAS and PI3K is what sends out the uncontrolled growth signals. If that handshake could be blocked, the signal would be cut off without shutting down PI3K’s other necessary functions, like helping regulate insulin.
To make this work, they used Vividion’s unique chemoproteomics platform, which scans huge libraries of molecules to find new drug targets that traditional methods might miss. Using this technology, they discovered a way to bind a molecule to a specific spot on PI3K that prevents it from interacting with RAS. This binding site isn’t involved in PI3K’s normal functions, so the drug blocks the cancerous interaction while leaving the rest of PI3K’s healthy roles intact.
The real breakthrough here is the proof that chemoproteomics can uncover hidden druggable sites that were previously thought to be “undruggable.” This opens up new possibilities for targeting tough diseases that haven’t had viable treatments.
In mouse models with human RAS-mutated lung cancer, the compound (VVD-642) stopped tumor growth without causing the dangerous side effects seen with previous PI3K inhibitors.
Importantly, there were no signs of hyperglycemia, which was a major issue with earlier drugs. This showed that the drug could specifically target the cancerous mechanism without disrupting normal metabolism.
Cancer is known for finding ways around treatments, so combining VVD-642 with other drugs targeting different parts of the RAS signaling pathway produced even better results.
Tumor growth was more effectively suppressed and for a longer time when VVD-642 was used alongside MAPK pathway inhibitors or direct RAS inhibitors like sotorasib.
This suggests that VVD-642 could play a key role in combination therapies, preventing the cancer from adapting and resisting the treatment. These findings are now shaping the design of clinical trials to test these combinations in humans.
You May Also Like: Study Finds 8 Powerful Lifestyle Habits That Could Add 20+ Years to Your Lifes (And They’re Easier Than You Think)
Expanding the Battlefield: The Unexpected HER2 Connection
What started as a targeted therapy for RAS-driven cancers has unexpectedly evolved into a broader cancer-fighting tool. When the researchers tested their new PI3K interaction inhibitor against cancers driven by HER2, they discovered it worked just as effectively.
This surprise finding opened up new possibilities, turning what was initially a specialized drug into a more versatile treatment.
HER2 (Human Epidermal Growth Factor Receptor 2), a well-known cancer driver, is responsible for pushing cells to grow uncontrollably. It’s typically overexpressed in about 15-20% of breast cancers, leading to aggressive, fast-growing tumors. While it’s most famous for its role in breast cancer, HER2 also plays a part in certain ovarian, bladder, and gastric cancers.
In these cancers, HER2 proteins form dimers, pairs that trigger a cascade of signals inside the cell, including activating the PI3K pathway. This makes PI3K a crucial player in both HER2-driven cancer and RAS-driven cancer, as both rely heavily on PI3K for their pro-growth signals.
The breakthrough came when researchers tested VVD-642 in HER2-positive mouse models. To their surprise, the drug not only worked but blocked tumor growth through a mechanism unrelated to RAS mutations. This discovery showed that the drug could also target HER2-driven cancers by disrupting PI3K activation.
By targeting this shared PI3K pathway, the drug can effectively hit two major oncogene families, RAS and HER2, giving it the potential to treat a variety of cancers. It works not by attacking a specific mutation but by interfering with a common cancer-driving mechanism.
The drug targets the PI3K “gate,” preventing both RAS and HER2-driven cancers from getting through. And because the drug targets a shared mechanism, there’s the potential for it to be effective against other cancers driven by different oncogenes that rely on PI3K activation, opening up even more therapeutic possibilities.
The leap from promising preclinical data to real-world human treatment is a long and meticulous journey, beginning with clinical trials. The recent success of the RAS-PI3K inhibitor has brought it to a crucial milestone: entering human testing through Phase 1 trials.
What is a Phase 1 Trial?
A Phase 1 trial is the first time an experimental drug is administered to humans. While it might seem intuitive that this phase is meant to evaluate the drug’s effectiveness, the primary focus is actually on safety and understanding how the drug behaves in the body. Key objectives of Phase 1 trials include:
Safety and Tolerability: Identifying any potential side effects (adverse events) that could arise from the drug and evaluating their severity and frequency.
Dose Finding: Determining the optimal dosage. Typically, the trial follows a dose-escalation design, where participants receive progressively higher doses until the Maximum Tolerated Dose (MTD) is found, the highest dose that doesn’t cause unacceptable side effects.
Pharmacokinetics (PK): Understanding how the body absorbs, distributes, metabolizes, and eliminates the drug.
Phase 1 trials usually involve a small number of participants, often those with advanced cancers who have exhausted other treatment options. This phase is an essential step in determining whether a drug is safe enough to move forward in the clinical trial process.
The VVD-642 Trial (NCT06804824)
The novel RAS-PI3K inhibitor, VVD-642 (also known as VVD-159642), has officially entered a Phase 1/1b clinical trial, registered under the identifier NCT06804824.
This open-label, multi-center study aims to evaluate the drug’s safety, tolerability, and early signs of efficacy in patients with advanced solid tumors that have been confirmed to harbor either a RAS alteration or HER2 amplification.
The trial design is informed by promising preclinical data and includes several arms to comprehensively assess the drug:
Monotherapy Dose Escalation: This arm is focused on establishing the safety and Recommended Phase 2 Dose (RP2D) of VVD-642 when given alone.
Monotherapy Dose Expansion: Once the RP2D is established, this arm will explore the safety and preliminary anti-tumor activity of the drug in specific patient groups.
Combination Therapy Arms: These arms aim to assess the safety and initial activity of VVD-642 in combination with other therapies. Specifically, it will be tested alongside KRAS-G12C inhibitors (like sotorasib) and MEK inhibitors (like trametinib).
The Dual Blockade Approach

What makes this trial especially noteworthy is its inclusion of combination therapy arms right from the start. The research team hypothesizes that VVD-642 will be most effective when used in tandem with other drugs targeting the MAPK pathway or mutant RAS itself.
By blocking both the PI3K pathway (with VVD-642) and the MAPK pathway (with trametinib) or directly targeting mutant RAS (with sotorasib), the trial is testing a dual blockade approach.
This combination strategy is theoretically powerful because it targets the two main arms of RAS signaling, potentially overcoming challenges like drug resistance.
What makes this trial even more exciting is the possibility that it may deliver this dual blockade more tolerably than older drug combinations, which often had high toxicity.
Therefore, this trial is not only testing a new drug but also exploring a potentially more effective and safer way to tackle RAS-driven cancers.
The Road Ahead: What Happens Next?
The initiation of a Phase 1 clinical trial for VVD-642 is an exciting and crucial milestone, but it is just the beginning of a much longer and more intricate journey. If the drug successfully proves to be safe and well-tolerated during Phase 1, it will then move on to subsequent stages of clinical development.
Phase 2: These trials involve a larger group of patients (typically 100-300) and are designed to get a preliminary assessment of the drug’s efficacy in a specific cancer type. They also continue to gather more safety data.
Phase 3: If Phase 2 results are promising, the drug moves to large-scale Phase 3 trials, which can involve several hundred to several thousand patients. These trials are typically randomized and controlled, comparing the new drug against the current standard of care to determine if it is more effective.
FDA Approval: Only after successfully completing all three phases and demonstrating a favorable benefit-risk profile can a company submit a New Drug Application (NDA) to regulatory bodies like the U.S. Food and Drug Administration (FDA) for approval to market the drug.
This entire process, from the start of Phase 1 to potential approval, can easily take five to ten years or more. The exciting news so far is just the beginning of a much larger story in the fight against RAS-driven cancers and potentially beyond.